



















































AI của Deepmind "thay đổi cuộc chơi" giải cấu trúc protein
Ngày đăng: 07/12/2020 09:58
Ngày đăng: 07/12/2020 09:58
Trí tuệ nhân tạo (AI) đã giải quyết một trong những thách thức lớn của sinh học: dự đoán cách các protein cuộn lại từ một chuỗi axit amin tuyến tính thành hình dạng 3D để thực hiện chức năng.
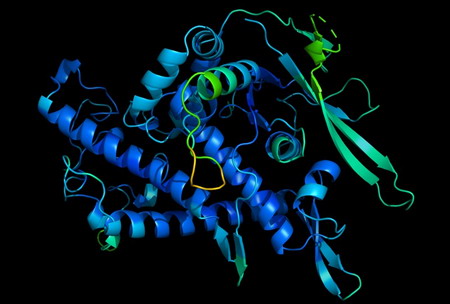 |
| Chức năng của protein được xác định bởi hình dạng 3D của nó. |
AI này do các nhà nghiên cứu tại DeepMind có trụ sở tại Vương quốc Anh, phát triển và dự kiến sẽ có những tác động sâu rộng, tăng tốc đáng kể việc tạo ra các loại thuốc mới.
"Những gì mà nhóm DeepMind đã đạt được thật tuyệt vời và sẽ thay đổi tương lai của nghiên cứu sinh học cấu trúc và protein," Janet Thornton, giám đốc danh dự của Viện Tin sinh học Châu Âu, bình luận.
Cuộc đua cuộn protein
Cơ thể con người sử dụng hàng chục nghìn loại protein khác nhau, mỗi loại có hàng chục đến hàng trăm loại axit amin. Thứ tự của các axit amin quyết định vô số cách đẩy và kéo giữa chúng làm phát sinh các hình dạng 3D phức tạp của protein, từ đó xác định cách protein hoạt động. Biết được những hình dạng đó giúp các nhà nghiên cứu tạo ra các loại thuốc có thể nằm trong các túi và đường nứt của protein. Và dự đoán được cấu trúc sẽ giúp tổng hợp nên protein với cấu trúc mong muốn, tăng tốc độ phát triển các enzym tạo nhiên liệu sinh học và phân hủy nhựa phế thải.
Trong nhiều thập kỷ, các nhà nghiên cứu đã giải mã cấu trúc 3D của protein bằng cách sử dụng các kỹ thuật như tinh thể học tia X hoặc kính hiển vi điện tử lạnh (cryo-EM). Nhưng những phương pháp như vậy có thể mất vài tháng hoặc vài năm và không phải lúc nào cũng hiệu quả. Đến nay, các nhà khoa học mới giải được cấu trúc của khoảng 170.000 trong số hơn 200 triệu protein được phát hiện ở các dạng sống.
Vào những năm 1960, giới ghiên cứu nhận ra rằng nếu tìm ra tất cả các tương tác riêng lẻ trong chuỗi protein, họ có thể dự đoán hình dạng 3D của nó. Tuy nhiên, với hàng trăm axit amin trên mỗi protein và nhiều cách mà mỗi cặp axit amin có thể tương tác, số lượng cấu trúc có thể hình thành trên mỗi trình tự là rất lớn. Các nhà khoa học tính toán từ lâu đã nghiên cứu vấn đề này, nhưng tiến độ rất chậm.
Năm 1994, John Moult, nhà sinh vật học cấu trúc tại Đại học Maryland và các đồng nghiệp tổ chức cuộc thi dự đoán cấu trúc cuộn protein CASP, cứ 2 năm một lần. Người tham gia nhận được trình tự axit amin của khoảng 100 protein chưa biết cấu trúc. Sau đó, ban tổ chức CASP so sánh các dự đoán cấu trúc mà các nhóm đưa ra với kết quả trong phòng thí nghiệm, và chấm điểm các dự đoán dựa trên độ trùng khớp với kết quả thí nghiệm (từ 0 - hoàn toàn khác, đến 100 - hoàn toàn trùng khớp). "Điểm dự đoán [hay gọi là điểm GDT] cao hơn 90 trên thang điểm từ 0 đến 100 được coi là ngang bằng với các phương pháp thực nghiệm," Moult nói.
Từ năm 1994, dự đoán cấu trúc cho các protein nhỏ, đơn giản có thể khá trùng khớp với kết quả thí nghiệm. Nhưng đối với các protein lớn hơn, điểm GDT chỉ vào khoảng 20, "hoàn toàn thảm họa", Andrei Lupas, giám khảo CASP và nhà sinh học tiến hóa tại Viện Sinh học Phát triển Max Planck, cho biết. Đến năm 2016, có nhóm đã đạt được 40 điểm khi dự đoán cấu trúc các protein "khó nhằn" nhất, chủ yếu bằng cách tái sử dụng thông tin từ các cấu trúc protein đã biết trước đây và có liên quan chặt chẽ đến các protein mà CASP đưa ra.
“Thay đổi cuộc chơi”
DeepMind tham gia thi CASP lần đầu tiên vào năm 2018, sử dụng thuật toán gọi là AlphaFold. Cũng dựa trên chiến lược đối chiếu với các protein đã biết như vậy, nhưng AlphaFold kết hợp thêm với học sâu hay phần mềm được đào tạo trên kho dữ liệu khổng lồ về trình tự và cấu trúc của các protein đã biết và học cách phát hiện ra các mẫu hình lặp lại. Năm 2018, DeepMind đã chiến thắng một cách dễ dàng, đánh bại các nhóm đối thủ với điểm trung bình cao hơn các nhóm khác khoảng 15% trên mỗi cấu trúc protein, và đạt đến 60 điểm GDT khi dự đoán cấu trúc các protein khó nhất.
Nhưng John Jumper, người đứng đầu bộ phận phát triển AlphaFold tại DeepMind, cho biết những dự đoán của thuật toán khi đó vẫn còn quá thô và chưa hữu ích. "Chúng tôi biết còn xa mới đến được các tiến bộ sinh học," Jumper nói. Để cải thiện, Jumper và các đồng nghiệp đã kết hợp học sâu với “thuật toán căng thẳng” - cho AI bắt chước cách một người lắp ráp một trò chơi ghép hình: trước tiên kết nối các mảnh thành các các cụm nhỏ - trong trường hợp này là các cụm axit amin - và sau đó mới tìm cách để nối các cụm thành một tổng thể lớn hơn. Họ đã đào tạo AlphaFold trên tất cả 170.000 cấu trúc protein đã biết.
Cải tiến này đã có tác dụng. Trên các protein mục tiêu trong CASP 2020 năm nay, AlphaFold đã đạt được điểm dự đoán GDT trung bình là 92,4. Đối với các protein khó và phức tạp nhất, AlphaFold đạt điểm trung bình là 87, vượt nhóm về nhì hẳn 25 điểm. AlphaFold thậm chí còn đạt kết quả xuất sắc trong việc giải các cấu trúc của protein nằm trong màng tế bào, vốn là trung tâm của nhiều bệnh ở người nhưng rất khó giải cấu trúc bằng phương pháp tinh thể học tia X. Kết quả này là “một bước tiến đáng kinh ngạc về vấn đề cuộn protein”, theo Venki Ramakrishnan, nhà sinh học cấu trúc tại Phòng thí nghiệm Sinh học Phân tử thuộc Hội đồng Nghiên cứu Y học Anh.
Moult nói rằng tất cả các nhóm trong cuộc thi năm nay đều tiến bộ. Nhưng AlphaFold, theo Lupas, "đã thay đổi cuộc chơi". Nhà tổ chức thậm chí còn lo rằng DeepMind có thể đã gian lận bằng cách nào đó. Vì vậy, Lupas đã đặt ra một thử thách đặc biệt: giải cấu trúc một protein màng từ một loài vi khuẩn cổ. Trong 10 năm, nhóm Lupas đã thử mọi thủ thuật tinh thể học tia X để giải cấu trúc protein này nhưng không thành công, họ có kết quả X quang nhưng chưa thể đọc ra hình ảnh cấu trúc từ đó.
Nhưng AlphaFold không gặp khó khăn gì. Nó trả về một hình ảnh chi tiết, cho phép nhóm Lupas hiểu được dữ liệu X quang sẵn có; dữ liệu trùng khớp với cấu trúc AlphaFold dự đoán. “[Trùng khớp] gần như hoàn hảo,” Lupas nói. "Không thể có chuyện gian lận."
Theo điều khoản tham gia CASP, DeepMind - giống như tất cả các nhóm khác - đã đồng ý chia sẻ đầy đủ chi tiết về phương pháp của mình để các nhóm khác sử dụng. Tiến bộ của AlphaFold sẽ rất có lợi cho các nhà thực nghiệm. Họ có thể sử dụng các dự đoán cấu trúc chính xác để hiểu dữ liệu tia X và cryo-EM đã có nhưng chưa thể đọc được, như của Lupas. Ngoài ra, phương pháp mới còn cho phép các nhà thiết kế thuốc nhanh chóng tìm ra cấu trúc của mọi protein trong các mầm bệnh mới và nguy hiểm như SARS-CoV-2, một bước quan trọng trong việc tìm kiếm các phân tử ngăn chặn bệnh, Moult nói.
Tuy nhiên, AlphaFold vẫn chưa thể làm tốt mọi thứ. Trong cuộc thi, nó đã chùn bước đáng kể trước một protein, hỗn hợp của 52 phân đoạn nhỏ lặp lại, làm sai lệch vị trí khi chúng cuộn thành protein. Jumper cho biết nhóm nghiên cứu hiện muốn đào tạo AlphaFold để giải các cấu trúc như vậy, cũng như cấu trúc của các phức hợp protein hoạt động cùng nhau để thực hiện các chức năng chính trong tế bào.
Theo Khoahocphattrien